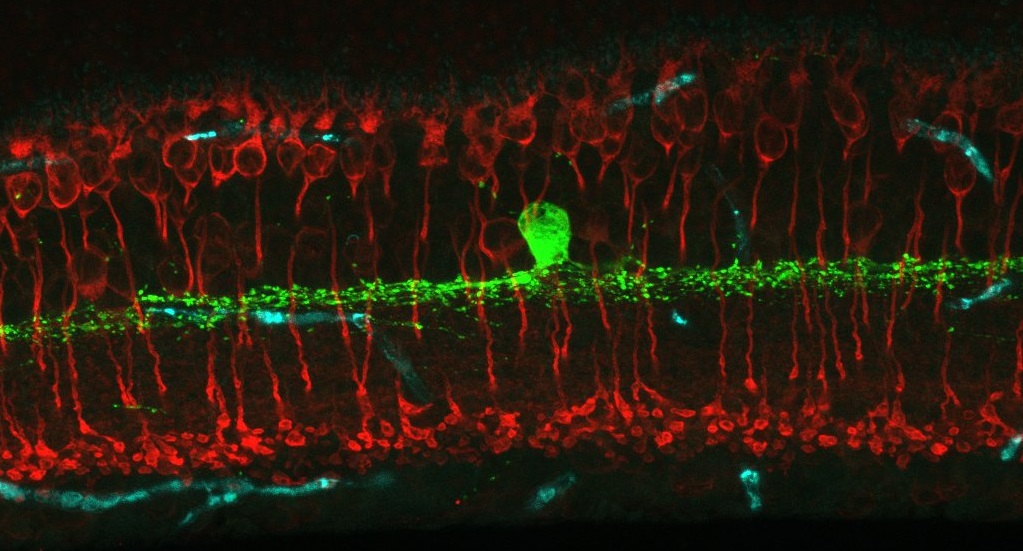
Welcome to the Eggers Laboratory of Retinal Neurophysiology! We are interested in modulation of retinal synaptic signaling by lighting conditions and disease – especially early diabetes. To investigate this we use many techniques including single-cell electrophysiology, immunohistochemistry, fluorescent in-situ hybridization and in vitro electroretinogram recordings (ERG).